Protein Epitope Prediction
Developed a bioinformatics pipeline integrating AlphaFold2 and transformer models for CD4+ T-cell epitope prediction in the Gragert Lab.
← HomePredicted CD4+ T-cell Epitope Likelihood
0 = Low, 1 = High (for antigen processing & MHCII binding)
Background
Antigen Presentation and the Need for Better Epitope Prediction
CD4⁺ T-cells inspect peptides displayed by MHC class II molecules and then coordinate downstream immune responses—responses that can be protective (vaccination) or harmful (graft rejection). Most public prediction servers still center on peptide–MHC binding affinity alone (e.g., NetMHCII; Nielsen & Lund 2009), implicitly assuming every overlapping peptide in a protein has an equal chance of reaching the MHC groove. In reality, endo-lysosomal proteases destroy the majority of sequence space before binding can even occur (Landry 1997; Mettu et al. 2016). Ignoring this “up-stream” filter inflates false-positive rates and limits the practical value of affinity-only models in vaccine design and HLA mismatch risk scoring.
Structural Constraints: From Crystal B-factors to Antigen-Processing Likelihood (APL)
Protease susceptibility tracks strongly with local backbone flexibility—captured in crystal structures by temperature factors, or B-factors—as well as with solvent exposure, COREX stability scores, and sequence entropy (Hubbard et al. 1994; Hilser & Freire 1996). The Antigen-Processing Likelihood (APL) framework (Mettu et al. 2016; Charles et al. 2022) fuses these four signals into a residue-level score that marks cleavage hotspots and, by extension, the adjacent stable stretches most likely to surface as CD4⁺ epitopes. Historically, however, APL has relied on experimental B-factors from X-ray structures—a requirement that excludes > 80 % of the proteome and blocks rapid analysis of novel vaccine antigens or rare HLA alleles.
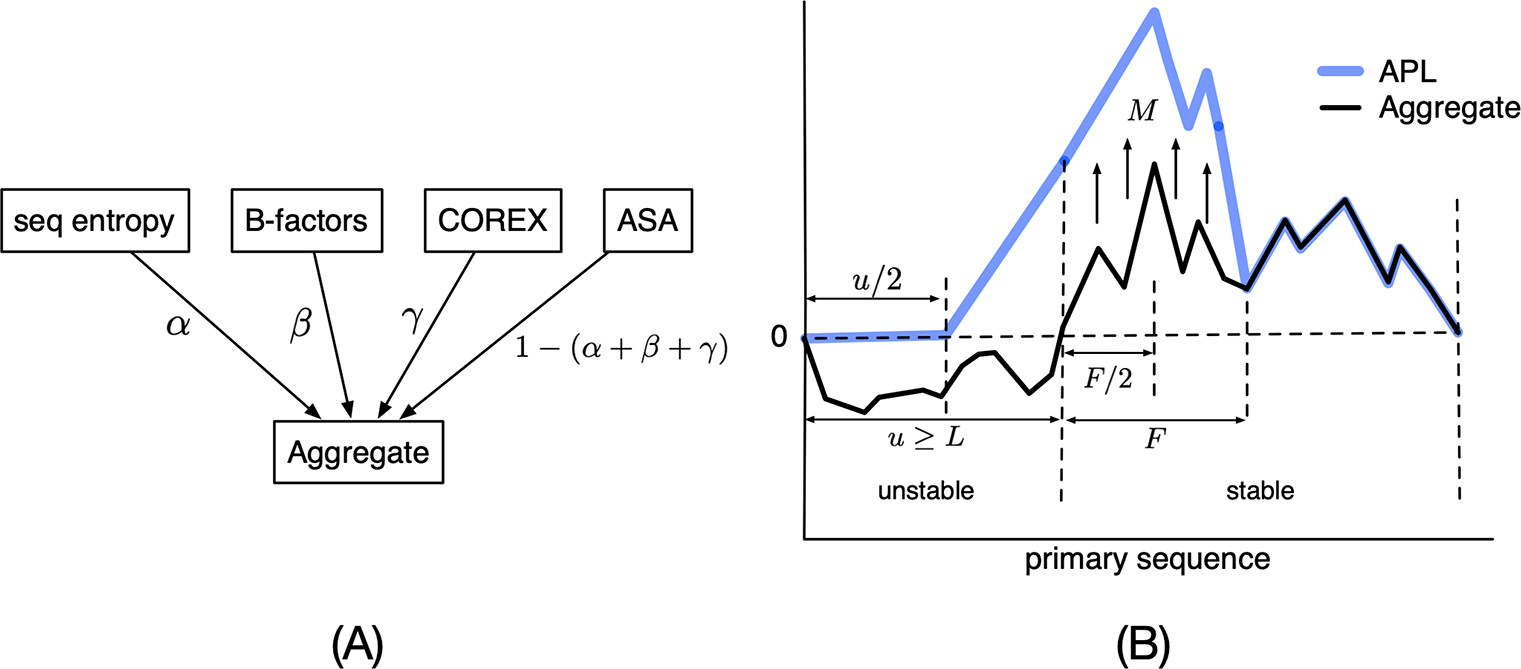
How APL (Antigen Processing Likelihood) Integrates Protein Structure Signals for Epitope Prediction
Panel A: the APL (Antigen Processing Likelihood) framework integrates four independent indicators of local flexibility and disorder—sequence entropy, crystallographic B-factors (or learned substitutes in our pipeline), COREX stability scores, and solvent-accessible surface area (ASA)—into a weighted aggregate. The weighting parameters (α, β, γ, etc.) allow each signal's contribution to be tuned based on predictive power for antigen processing.
Panel B: the model interprets the aggregated structural profile along a protein sequence. Unstable regions (high flexibility) are assigned low scores, reflecting high protease susceptibility and low epitope likelihood. In contrast, stable regions adjacent to these flexible zones are "upweighted" by a magnification factor (M), marking them as probable sites where proteolytic processing will generate peptides likely to survive for presentation in the MHCII groove. The resulting APL score (blue line) effectively highlights regions with high predicted CD4+ T-cell epitope likelihood, capturing the reality that not all peptides are equally likely to reach the cell surface.
Mettu, Ramgopal R., Samuel J. Landry, Tysheena Charles, Daniel L. Moss, Pawan Bhat, Peyton W. Moore, Nicholas A. Kummer, and Avik Bhattacharya. "CD4+ T-cell epitope prediction by combined analysis of antigen conformational flexibility and peptide-MHCII binding affinity." Biochemistry, vol. 61, no. 15, 2022, pp. 1585–1599. https://doi.org/10.1021/acs.biochem.2c00237.
In my work, this framework is adapted for high-throughput prediction by replacing experimental B-factors with accurate, learned flexibility scores from our AlphaFold2 + transformer pipeline—making APL accessible for any protein, including those lacking experimental structures. Integrating these structure-aware predictions with state-of-the-art MHCII binding models enables more precise, end-to-end ranking of potential CD4+ T-cell epitopes for vaccine and immunogenicity applications.
Removing the Structure Bottleneck with Learned B-factor Prediction
AlphaFold2 has democratized access to near-atomic protein coordinates but does not output B-factors, leaving a critical gap for structure-aware epitope prediction. To bridge it, I first trained a transformer on the entire UniRef sequence corpus with a masked-token auto-encoding objective, generating context-rich embeddings that far outperform one-hot encodings for downstream tasks. I then combined these embeddings with geometric features extracted from AlphaFold2 models in a new distillation network that regresses crystallographic B-factors, achieving a Pearson correlation > 0.8 to experimental values. These learned flexibility scores, together with AlphaFold2 geometries, allow me to compute all four APL inputs for any protein—no crystal structure required.
By coupling learned flexibility scores derived from AlphaFold2 models with predicted MHC-II binding affinities, the updated APL workflow operates entirely in silico. It ranks CD4⁺ T-cell epitope likelihoods directly from protein sequences, providing a structure-aware tool for immunogenicity assessment across diverse antigens.
Contributions
- Improved protein sequence embeddings via masked-token prediction and AlphaFold2 distillation for b-factor prediction (PCC>0.8).
- Developed a bioinformatics pipeline combining AlphaFold2 and transformer models for CD4+ T-cell epitope prediction.
- Architected and deployed a web server and standalone tool for immunology and bioinformatics analysis.
- Built a distributed GPU-accelerated RESTful API framework, reducing compute time by up to 180x.
- Published technical reports and led cross-functional discussions to refine epitope prediction algorithms.